{kind=link}
{kind=link}



![]()
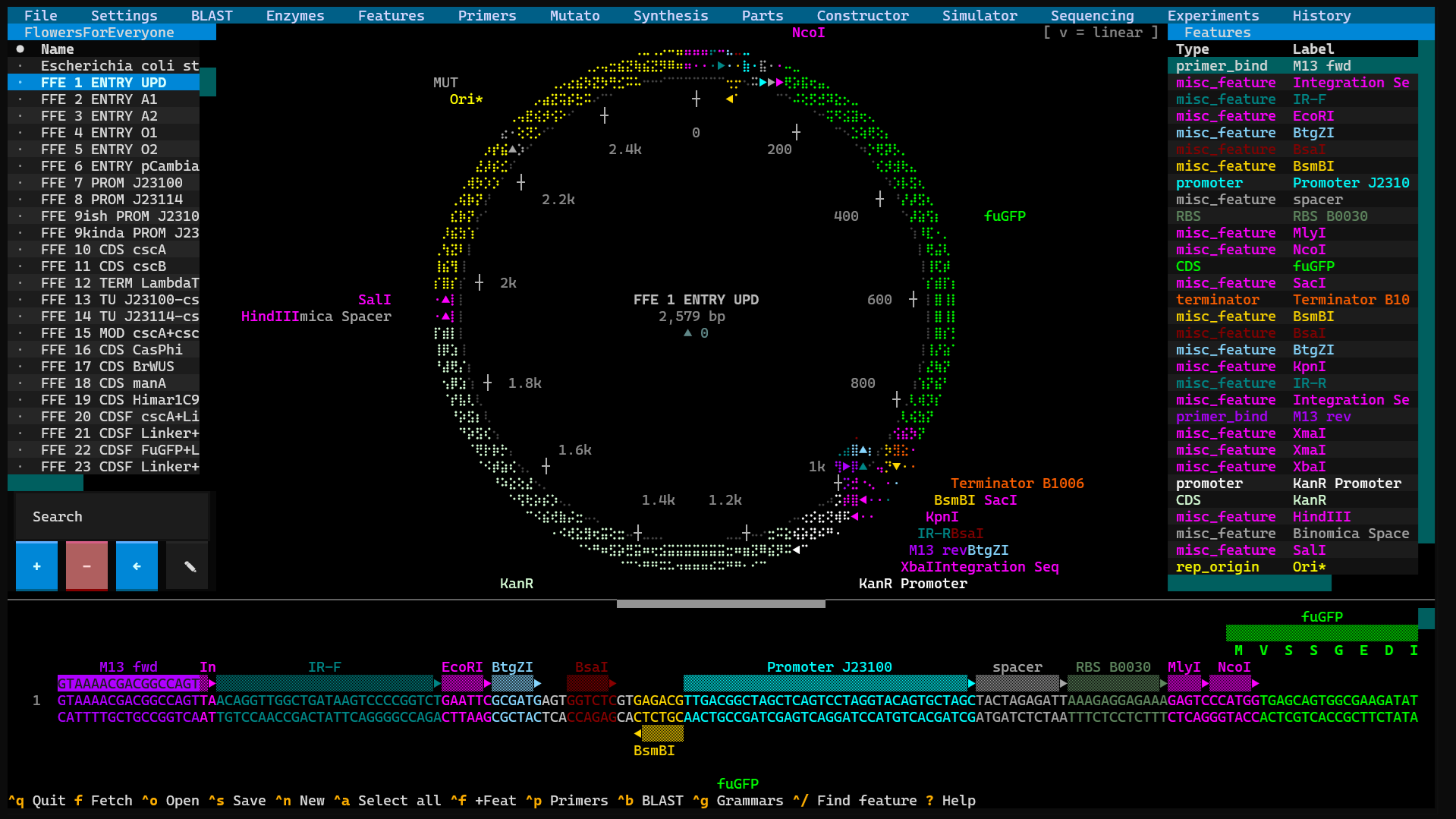
SpliceCraft is a plasmid workbench that runs where you already work. Open a map, edit the sequence, design primers, plan a Golden Braid or MoClo assembly, BLAST a hit, check your Sanger reads, and keep a lab notebook — all from the keyboard, in one place, no browser tab and no cloud account. Circular and linear maps render as crisp Unicode braille graphics in any modern terminal, and nothing leaves your machine unless you ask it to — and any plasmid's map exports to a publication-quality PNG or SVG (with a transparent-background option for figures), one at a time or a whole batch straight from the library.
It's built by a practicing bioengineer for daily bench work: the bug reports come from real cloning, and so do the fixes.
Why give it a try:
- Fast and local. No Electron, no web app, no login.
pipx install splicecraftand you're designing in seconds. - It does the whole job. View → edit → design → clone → simulate → verify → document — one tool that understands how those steps connect.
- It guards your data like it's irreplaceable (because it is — see below).
- It's scriptable. A 150+ endpoint local API and a stdlib CLI let an agent or a shell script drive every workflow.
pipx install splicecraft
splicecraft # empty canvas
splicecraft L09137 # fetch pUC19 from NCBI on launch
splicecraft myplasmid.gb # local GenBank or .dnax86-64 Linux, Intel macOS, and Windows install entirely from prebuilt
wheels — nothing to compile. On ARM64 Linux (Raspberry Pi / ARM cloud)
and Apple Silicon, one dependency (primer3-py) has no ARM wheel and
compiles at install, so install a C toolchain first:
sudo apt install build-essential python3-dev (Linux) or
xcode-select --install (macOS), then pipx install splicecraft.
On Windows, run SpliceCraft in Windows Terminal (the Windows 11
default) for the braille map — it auto-configures the console for UTF-8 +
ANSI at startup, so the map should render instead of garbling. (This
native-Windows path is implemented and CI-tested via mocks, but not yet
confirmed on real Windows hardware — see docs/PLATFORMS.md.)
If braille shows as
boxes (a font without those glyphs), toggle 'ASCII plasmid map' in
Settings → Display. See docs/PLATFORMS.md for the full
terminal matrix.
Press ? once running for the full keyboard-shortcut reference, or Ctrl+K for a
fuzzy command palette that jumps straight to any tool by name. See
docs/install.md for pip / uv / conda / source installs.
Your plasmid library is months — sometimes years — of work, so SpliceCraft is built to be a daily driver you never have to worry about:
- Your data is sacred. Every save is atomic (a crash can't leave a half-written file), backed up (
.bak+ rotating timestamps + daily snapshots), and guarded by a "suspicious shrink" refusal that won't replace a 156 MB library with an empty file. Name collisions always ask — skip / copy / overwrite — and self-updates snapshot everything first. - The biology is correct, and proven. Palindromes, Type IIS, origin-spanning cuts, wrap-around features, non-standard genetic codes (
/transl_table), reverse-complement, and IUPAC are pinned to the base — behind 4,000+ tests plus property-based fuzzing on the biology, crash-injection on the save path, and concurrency fuzzing on the data layer. Releases ship only when the whole suite is green. - We go looking for trouble. A long list of "sacred invariants" (
CLAUDE.md) and deep, multi-pass pre-release audits hunt edge cases, data-loss windows, races, and security gaps before they reach you.
Data-safety writeup: docs/data-safety.md ·
Security policy: SECURITY.md.
Everything hangs off a menu bar across the top, read left to right. The full
reference lives in docs/features.md; here's the gist.
Search without leaving the app (Ctrl+B). Local runs BLASTN / BLASTP /
HMMscan against your own library in-process — powered by pyhmmer, so there's
no external blast+ to install — with a one-click Pfam-A / NCBIfam (or any
HMMER3 URL) downloader. Online sends DNA / protein — or a whole plasmid or
single feature — to NCBI or EMBL-EBI Pfam and tables the hits, with a live
poll counter and a Cancel that really stops. Add to collection pulls a
highlighted nucleotide hit's full GenBank record straight from NCBI into a
plasmid collection you pick. (Native Windows: HMMscan needs WSL2; BLASTN/BLASTP
run in-process.) Scripting agents can run online search too — but only once
you tick Settings → "Allow agent online BLAST/HMMER", so a script or an
autonomous assistant can never silently ship your sequences off-box; the
in-process BLAST/HMMscan they always have stays on your machine.
Drive the restriction overlay — all sites, unique cutters, 6+/4+ bp, or just the Golden Braid connectors. Multi-cutters wear a live superscript cut-count (EcoRI², BsaI³) that ticks down as you edit a site out. Build named enzyme collections from the 200+ NEB catalog plus your own customs; the active collection scopes every scan. Your custom enzymes are now offered in every enzyme picker — the overlay list, the traditional-cloning and Golden-Braid / MoClo choosers — and the cloning-grammar editor accepts them too.
A library for your reusable annotations — promoters, RBSs, tags, CDSs. Capture
a region off any plasmid, then drop it onto another to annotate a selection
or splice the sequence in (the same store Synthesis and the Domesticator
use). Ctrl+F finds a subsequence — fuzzy, both strands — and n/N step
the hits, each pre-selected so Alt+Shift+F tags it on the spot. (Ctrl+/
searches features by name instead.)
A full-screen Primer3 designer for detection, cloning, Golden Braid, and generic primers, each with a Designed → Ordered → Validated lifecycle shown beside its plasmid. A fifth Primer Check tab runs in-silico PCR across your library (or just the active collection): one primer lists every plasmid it anneals to with the % identity, strand, and position; two primers add the amplicon length and the feature amplified, ranked by confidence (✓ / ⚠ / ~ / ✗). Binding is judged on the primer's 3′ end, so a 5′ cloning tail shows as lower identity rather than vanishing — click a result to open it on the canvas at the binding site.
The primer library organises into collections with a fuzzy search bar. Space cycles a primer's mark (★ select · $ cart · M move); MOVE / bulk-delete / re-status the marked sets, and export a collection or your $ cart to an order-ready CSV — generic (Name/Sequence/Length/Tm) or the IDT bulk-upload template (Name/Sequence/Scale/Purification) — (then import one back). Marks track the primer itself, so filtering never strays them; malformed oligos are refused on export and skipped on import. Ctrl+C copies the highlighted primer's sequence (with a base-count toast).
Site-directed mutagenesis (with a hint of whimsy). Point at a CDS, name the
change (L54A), and SpliceCraft designs the SOE-PCR primers — falling back to a
2-primer modified-outer strategy near the ends, and only offering the shortcut
when the primer genuinely carries the change, so you never amplify wild-type by
accident. It also turns a pasted protein into a ready-to-order CDS:
frequency-matched codon optimization against your table, a stops selector
(1–3, honoring a trailing * run), and an Avoid sites picker that scrubs
chosen cut sites out of the CDS.
Its Scrub tab cures a whole plasmid of restriction sites with no cloning:
pick the enzymes (Type IIS by default) and SpliceCraft finds the minimal point
changes that kill each site — silent across every overlapping reading frame,
never spawning a new site, and reported when a site can't be cured silently.
Apply cure names and saves the cured plasmid (primers bound where they
anneal, drawn on the original as mismatches) and re-circularizes by
QuikChange (PCR → DpnI) or Golden Braid (BsaI-tailed fragments ligated
back together) — the Golden Braid route saves each PCR-… amplicon and really
digests + ligates them, so History reads as a genuine assembly.
A gene-synthesis composer in three tabs (the main menu bar stays on-screen so you can switch straight to another tool and back — switching closes the tool you're leaving rather than stacking screens, and each keeps its work; no save prompt on the way out, and Clear is the explicit per-tab wipe):
- DNA — a scrolling linear editor with anti-parallel strand markers, feature stripes, and live AA translation, plus a feature-library side-pane (insert / annotate) and a feature-aware paste (copy a plasmid stretch and its features ride along). Just like the main sequence viewer, click a feature stripe to highlight the whole feature and double-click (or press Enter inside it) to edit its label / type / color / notes, press R to reveal restriction cut sites — then click an enzyme name to highlight its recognition site plus, for Type IIS cutters, the spacer and staggered cut — extend a selection with Shift/Ctrl+click or Shift+arrow and nudge it with Ctrl+←/→, Ctrl+C to copy the selection byte-for-byte with its features, and Ctrl+Z / Ctrl+Y to undo / redo any typed bases or edits. Press −/+/0 to zoom the whole fragment out to a compact feature-block overview (with a bp ruler) and back — click any column to jump to 1:1 editing there. Pressing Delete on a highlighted feature removes just the annotation (press again to delete the highlighted bases). Insert Site stamps the enzyme's recognition sequence as a hot-pink, direction-arrowed feature — pick Forward ▶ or Reverse ◀ (reverse-complement) in the dialog.
- Protein — type or paste amino acids and watch codons fill in from your chosen table; a built-in motif library (His6, FLAG, HA, TEV, P2A, NLS, GS linkers, +30) inserts pre-colored tags, and pasted / inserted motifs are click-to-highlight, double-click-to-edit with the same Ctrl+Z / Ctrl+Y undo. Optimize → DNA codon-optimizes (with Stops auto-tracking the trailing
*run and the same Avoid sites scrubbing) and hands the CDS to the DNA tab. The tabbed codon-table manager (also at Settings ▸ Codon Tables) builds tables from an NCBI genome (highly-expressed genes or whole-genome), a local CDS file (cds_from_genomic.fna/.gz, fully offline), Kazusa, or TSV, and a Chart tab draws any table as the classic genetic-code grid. - Operon Design — Synthetic Operon Construction turns the codon optimizer + a built-in pure-Python RBS engine into an expression-tuning bench: drop proteins into a lane, give each a target relative RBS strength, and Assemble reverse-designs every RBS in its real assembled context (under-drivable genes flagged), dropping a fully-annotated operon into the DNA tab. Native Operon Domestication lifts a natural operon (canvas / library / NCBI), cures the grammar's forbidden Type IIS sites (plus any extras you list) with primer-encoded synonymous edits, and clones it in with features intact.
Compose a part, hit Clone Fragment, and pick a path: a modular grammar
hands it to the Domesticator as an L0 block; Gibson or Traditional
open the Constructor with it pasted in. Saving a domesticated part files
three things in one dialog — the cloned plasmid, the orderable linear
fragment (FRAG-…, the primed amplicon with its domestication primers +
features drawn on it), and the parts bin the L0 part lands in — each into any
collection. Nothing on your canvas is touched until you save.
Ordering the DNA synthetic instead of amplifying it? L0 Fragment wraps the
composed sequence in the correct nested overhangs for direct synthesis — pick a
grammar position (Promoter / CDS / Terminator …) or type a custom 4-nt overhang,
and the fragment comes back annotated and ready to order as a gBlock, so a
single Esp3I (BsmBI) cut drops it into your UPD / pUPD entry vector as a
proper L0 part. It's two-tier aware: when the configured entry vector's
external acceptor overhangs (e.g. CTCG/TGAG) differ from the part's category
overhangs (e.g. AATG/GCTT), the category pair is nested inside the
external pair automatically — the layout GoldenBraid needs so the part enters on
Esp3I and later releases on BsaI. Internal Type IIS sites in your insert are
flagged so you can scrub them (via Optimize) before ordering.
Your Parts Bin — the Level-0 building blocks for grammar-based assembly, in per-grammar bins. Multiple bins live side by side as collections, so a yeast toolkit and a plant toolkit never get mixed up.
The assembly bench: Traditional cloning, Gibson, Golden Braid, MoClo, or your own grammar, driven by a 4-source part picker. Every assembly, at every level, lands as one library entry (payload + overhangs + backbone) that carries every parent feature forward — so you can trace a finished L3 construct back to its L0 parts from the Library panel.
In-silico PCR and agarose gels. Pick a template, run the PCR, then save the
amplicon or send it to a gel lane. Gels render at 0.5–4% on a real
Helling–Goodman–Boyer mobility curve; stack lanes side by side, save a gel to
reload later, or cite it as &<gel> in your notebook.
Verify constructs against real reads. Drop in a Plasmidsaurus .zip — or
fetch a run by item code straight from the Plasmidsaurus API (the button on
the Sequencing screen; set credentials under Settings ▸ Plasmidsaurus API or via
the PLASMIDSAURUS_CLIENT_ID / PLASMIDSAURUS_CLIENT_SECRET env vars) — then walk
run → sample → target, and Align: the read lands as a colored bar (blue
match / red mismatch / gray gap) on the plasmid's linear map, named in place,
shaded by how much of each span actually binds so even a single-base mismatch
shows red. Click a read to jump the sequence panel to that exact spot.
Bulk auto-align matches a whole results folder in one pass, its confirm
window showing each read's real identity / mismatch / gap counts. The
Verification Report grades every construct (✓ verified / ⚠ near / ~ partial
/ ✗ divergent) in a sortable table; the Alignment Manager lists every stored
alignment (a true sub-100% identity never rounds up to "100%"); and the Library
shows per-plasmid Seq and Kind (○ plasmid · / fragment · ≈
amplicon · ρ protein) badges.
A genuine lab notebook in markdown: a split-pane editor, entries grouped into
projects (the way plasmids group into collections), and live colored
cross-references — type @plasmid, !action, or &gel and Ctrl+G jumps to
the source. Attach images — previewed inline in the notebook — and spellcheck
with F7 against a dictionary you can grow.
Every plasmid remembers how it was made — Golden Braid, digest/ligation,
Gibson, PCR, or a plain edit. History opens with a Protocol — a
numbered recipe that reads left → right like the bench ("assemble pProm +
pCDS_GFP + pTerm into pENTR_L1 → TU_GFP ✂ Esp3I") — above a lineage tree you
can drill into as deep as you like. Each step is dated and shows its detail
(including the primers for a PCR); a backbone reused across branches is shown
once and then referenced. The lineage rides along through CommercialSaaS .dna
import / export too.
A chat assistant that lives in the terminal next to your plasmids. BABS is a
direct conversation with a local Ollama model — no
cloud, no API key, nothing leaves your machine — wearing the Babs chat UX:
streaming answers with markdown highlighting, reasoning (<think>) hidden by
default, a ❤ context lifebar that shows how much chat memory is left, and
slash commands (/help, /model, /system, /temp, /reset, /retry,
/agent, /autonomy). The
transcript keeps a long, selectable, copy-pasteable history, and Ctrl+E
exports the whole conversation to markdown. Three tabs:
- Chat — ask anything; the answer streams in, contextually colored. Toggle
Corpus on (enabled once a Babs corpus exists) and answers come grounded
in your research corpus, with cited sources — the same hybrid retrieval Babs'
own
rag_botdoes, streamed into the chat. Off, it's a plain local-model chat. So you can grow a corpus in the Paper scraper and query it without leaving SpliceCraft. - Agent mode — flip Agent on (or
/agent) and Babs can drive SpliceCraft herself: she calls the same scripting endpoints the external--agentAPI exposes — read the loaded plasmid, find a motif, design primers, run a digest, clone, manage the library, even drive the OT-2 — and everything she does shows up live in the app. She's always told which plasmid you have open, so "domesticate this" or "what's in the construct" just work. By default she asks before every write (a one-tap approve/deny);/autonomy autolets her run unattended,/autonomy readonlykeeps her to read-only. Destructive whole-library wipes are never reachable, and physical robot motion always asks first — even inauto— so nothing moves the hardware unattended. She can carry out a whole multi-step workflow in one turn — a batch of endpoints run in order (inaskmode a single prompt lists every change, so you approve the batch once instead of click-by-click). A big local chat model can be slow in a multi-step tool loop, so agent turns automatically run on a fast, tool-capable model (qwen2.5:7b by default) while your chosen model still handles ordinary chat — override with/agentmodel <name>|chat|auto. And if you ask a lookup-type question with Agent off, Babs reminds you to turn it on so she can actually search instead of guessing from memory. - Online lookups — in Agent mode Babs can also look things up on the web: FPbase (fluorescent-protein spectra), UniProt (proteins), Europe PMC (papers), NCBI/GenBank (sequence records), Wikipedia, general web search, and patents — and she can open a result and read the page itself (the full text, not just the snippet; HTML stripped to text, no JavaScript, and PDFs/images are declined). Off until you tick Settings → "Allow Babs online database lookups" — and even then only your query string (or the URL you're reading) is sent, never your sequence (a local model has no internet on its own). Web + patent search use the free official APIs if you add a Brave Search / PatentsView key in Settings, otherwise fall back to keyless Google/DuckDuckGo (best-effort, rate-limited).
- Model — organize your models into collections, exactly like plasmids:
make/rename/delete collections, mark rows (Space) to move them between
collections or uninstall them in bulk, and file a model into a collection
before you've even pulled it. Every installed model is auto-filed into a
default "My Models" collection so nothing's hidden. Search HuggingFace for
GGUF models and add results to a collection, then pull on demand with a
live progress bar that shows bytes pulled and download speed (e.g.
1.2 GB / 5.5 GB · 21% · 18.4 MB/s) so you can see a multi-gigabyte model is really downloading. Use sets a model as the chat model. Deleting a marked model runsollama rm(behind a confirm — it frees disk and needs a re-pull). Everything runs over Ollama's local HTTP — no extra dependency. - Paper scraper — launch Babs'
real background paper search — Europe PMC plus OpenAlex, CORE, CGSpace and
DOAJ (all legitimately-open APIs) → re-index — to grow her knowledge corpus,
with a live, self-correcting "ingest running" indicator
and a jobs view + log tail. Switching models warns you (default No) that a
changed embedding model needs the corpus re-ingested to match, and can run
that re-embed for you. Shown only when the Babs repo is present (
~/babs, or$SPLICECRAFT_BABS_HOME). - Learn — grow Babs' knowledge about a specific topic with a focused,
drift-resistant crawl of open scientific databases (plus open-licensed web
pages). Give it a topic and a paper budget; it seeds from open-access search,
scores every candidate for relevance, and only follows citations out of the
strongly on-topic ones — so it deepens on your subject instead of wandering
off into adjacent fields. Each topic gets its own isolated corpus. Live status
(kept / fetched / dropped / frontier) and a scored "kept papers" table. Requires
arming Settings → "Allow Babs online database lookups" (only topic query strings
are sent — never your sequences). Also drivable over the agent API (
learn-start/learn-status/learn-results/learn-list). - Persistent memory — tell Babs something to keep with
/remember <fact>and it survives across sessions (loaded into every future conversation);/memoryshows what it holds. Stored as plain, hand-editable markdown in the Babs corpus, kept separate from crawled documents.
The BABS tab is persistent — switch to another part of SpliceCraft or close it and your conversation, model choice and agent mode are still there when you come back, and a model download you kicked off keeps running while you work elsewhere.
The toolbar scrolls horizontally when the terminal is too narrow to fit every
menu, so BABS at the far right is always reachable. Requires Ollama running
locally (ollama serve); point elsewhere with $SPLICECRAFT_OLLAMA_HOST. New to
it? Run splicecraft babs-setup to clone + bootstrap the Babs engine (venv,
deps, models) in one command, and the Chat tab shows a numbered first-run
checklist whenever Ollama or a model is missing.
File opens / fetches (NCBI) / saves / exports (GenBank · FASTA · GFF3 ·
circular-map image as PNG/SVG, one plasmid or a whole collection in
bulk), bulk-imports a folder, and restores from backup; every
GenBank it writes stamps
a traceable Created by SpliceCraft v… COMMENT. It also hosts the selection →
cloning hub (Alt+Shift+P): highlight any DNA and pick Traditional,
Golden Braid / MoClo, or Gibson — each opens pre-loaded with the
selection and its features. The Traditional branch steers you to a working
enzyme pair (flagging sites inside the selection, or that the vector can't open
with), designs the cut-site-tailed primers, saves the named amplicon, then on
Simulate digests and gel-purifies so no primer-pad bases leak into the
clone. Migrate Data packages your entire setup (library, collections, parts,
primers, features, grammars, codon tables, settings, notebook, and history) into
one checksum-verified .zip to move between machines, and Master Delete is a
triple-gated full wipe. Settings collects every toggle plus launchers for
the grammar, entry-vector, enzyme-collection, and codon-table editors.
Want to script all of this? A 150+ endpoint localhost JSON API
(splicecraft --agent, or --headless for a no-UI / no-pty server with a
/healthz readiness probe) and a stdlib-only CLI (splicecraft-cli,
including a call passthrough to every endpoint) drive every workflow.
/tools self-describes each endpoint's full request schema. See
docs/agent-api.md and docs/cli.md.
Full feature reference: docs/features.md.
SpliceCraft can also drive an Opentrons OT-2 liquid handler. The AUTOLAB
toolbar item is a five-panel protocol designer. Find Robots scans your network
(and USB) for OT-2s and lists what it finds in a pick-and-connect modal — or tick
Auto-connect to first to search and connect in one click. Then an interactive
Deck drawn as a top-down diagram of the robot — the slots as one connected grid
of boxes in the physical layout, each bay drawn at the real slot's rectangular
aspect ratio and the whole deck scaled + centred to your terminal, colour-filled by
what's on each bay (click a bay to place or clear its labware), a Designer for an ordered sequence of
steps (transfer, distribute, consolidate, mix, delay, pause, comment), a
Labware tab that keeps a library of custom labware you define from a simple
grid form, a Library tab that links the robot to your plasmid library —
bind a deck plate to a collection so its wells map to your plasmids, then
cherry-pick or replate them by identity, or normalise DNA concentration to a target
ng or ng/µL — and a Calibrate tab: read calibration status (deck + per-pipette
offset + tip-length), home the gantry, set per-slot labware offsets, and run a
position check that moves the gantry above each labware to verify alignment
(no aspirate/dispense — the plunger never actuates). Save whole designs as named
protocols organised into collections, compile to an Opentrons protocol, analyze on
the robot's built-in simulate, and run it (gated behind Arm + a clean analysis + a
pre-flight health + calibration + attached-pipette + door check) with live Pause /
Resume / Abort controls. A live telemetry panel tracks the robot as it works — a
connection badge, the robot's light / door / motor / pipette state, a run progress
bar with ETA, and a fault banner that pops the instant something goes wrong —
plus quick Lights On / Off and Disengage Motors buttons. A build can be logged
straight to the Experiments notebook, cross-linked to the plasmids it touched. All of
it is scriptable via the agent API — ot2-compile (a transfers list or a multi-step
steps sequence), ot2-analyze, ot2-status, ot2-calibration, the gated
ot2-run / ot2-position-check / ot2-home, ot2-run-control, ot2-lights,
ot2-disengage, ot2-normalize, ot2-plate-map, plus protocol- and custom-labware
library CRUD endpoints.
| Topic | Where |
|---|---|
| Install methods | docs/install.md |
| First five seconds with pUC19 | docs/getting-started.md |
| Full feature list | docs/features.md |
| Keybindings + menus | docs/keybindings.md |
| Data safety + backups | docs/data-safety.md |
| Agent API (HTTP) | docs/agent-api.md |
| CLI sidecar | docs/cli.md |
| Architecture | docs/architecture.md |
| Sacred invariants | CLAUDE.md |
| Contributing | CONTRIBUTING.md |
| Security policy | SECURITY.md |
| v1.0.0 acceptance gate | V1_GATE.md |
| Changelog | CHANGELOG.md |
| Release checklist | RELEASE_CHECKLIST.md |
python3 -m pytest -n auto -q # full suite (~5–6 min on 8 cores)
python3 -m pytest tests/test_dna_sanity.py # biology correctness only (< 2 s)
python3 -m pytest tests/test_perf_regression.py # perf gates (~3 s)All tests run offline against synthetic SeqRecords and monkeypatched data
paths; the autouse _protect_user_data fixture in tests/conftest.py
guarantees no test can write to real user files.
SpliceCraft is actively maintained by a practicing bioengineer running real cloning workflows in it daily; releases typically go out the same week a problem surfaces at the bench. Issues and PRs welcome at github.com/Binomica-Labs/SpliceCraft/issues.
See CONTRIBUTING.md before opening a non-trivial PR — it
walks through the sacred invariants, the test cadence, and the
security-sensitive code surfaces.
MIT